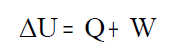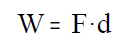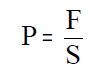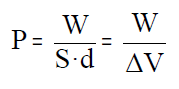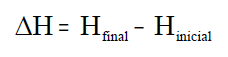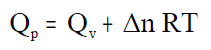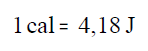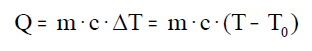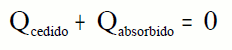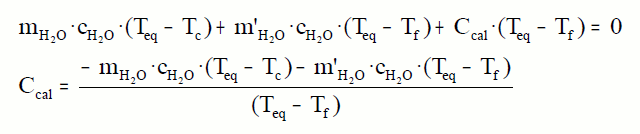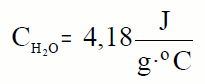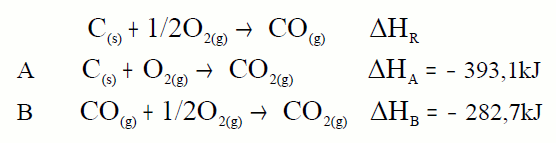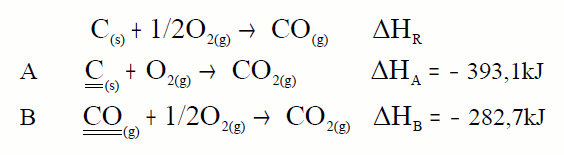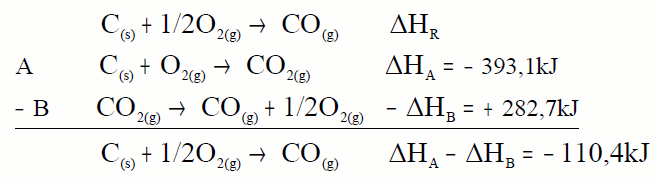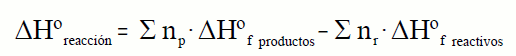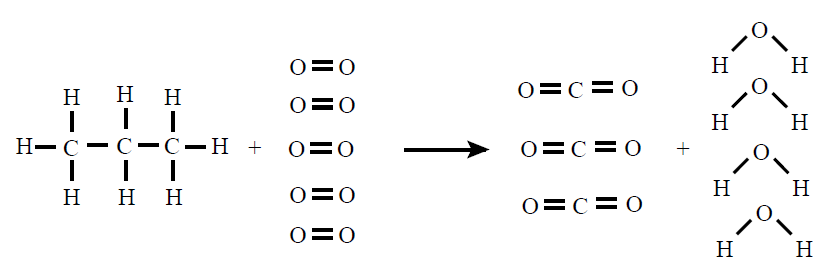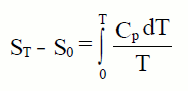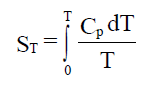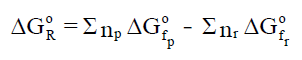|
|
|
|
OBXECTIVO DA TERMOQUÍMICA
|
| A Termoquímica é a aplicación da Termodinámica
ao estudo da química. A Termoquímica estuda os intercambios de enerxía que
acompañan ás reaccións químicas. É un feito experimental que en toda
reacción química hai unha variación de enerxía, manifestada normalmente pola
emisión ou absorción de calor. O obxectivo da Termoquímica é o estudo destas
variacións de enerxía (da súa importancia pode darnos idea o feito de que
non só hai moitas reaccións, en especial as de combustión, que teñen como
único obxectivo o aproveitamento da enerxía desprendida) e tamén o estudo da
espontaneidade dunha reacción.
Pódese dicir dunha maneira moi xeral que a termodinámica é
a ciencia da enerxía, e poderíase engadir que tamén se ocupa da
entropía.
A termodinámica está baseada, esencialmente en só dous postulados,
coñecidos como primeiro e segundo principios da termodinámica. Estes
axiomas non se deducen, en todo caso podémolos considerar
xeneralizacións de experiencias. Deles, por razoamentos lóxicos,
matemáticos, derívanse relacións entre as magnitudes macroscópicas, é
dicir, variables que se refiren a un gran conxunto de partículas
(volume, presión, temperatura, calores específicas, entalpía,...). A
validez destas relacións pódese comprobar experimentalmente, e con iso a
dos principios. Fai falta subliñar que a termodinámica é
unha teoría macroscópica, non fai falta ter en conta para nada a estrutura
atómica e molecular da materia. Esta característica confírelle unha gran
fiabilidade. Pode ser falsa a existencia dos átomos pero a termodinámica non
cambiará os seus principios, xa que non se deducen teoricamente senón da
experiencia. A termodinámica non describe o proceso de
cambio dos sistemas nin indica o tempo necesario para o mesmo, informando só
acerca da posibilidade de que exista tal cambio. A pesar de todo, saber si
unha reacción é posible ou non pode aforrar moitos esforzos inútiles, así,
se a termodinámica dixese que era imposible obter diamantes a partir de
grafito, a ninguén se lle ocorrería esforzarse tentándoo. Os
obxectivos da termoquímica podémolos resumir así:
- Estudo das variacións de enerxía, como entalpías de reacción,
enerxías libres...
- Saber, fixadas certas condicións (por exemplo a temperatura e as
concentracións), se é factible unha reacción ou non.
- Obtención, teórica, das constantes de equilibrio e métodos para
mellorar os rendementos das reaccións.
Con respecto a un proceso en concreto a termoquímica non
nos di nada da súa velocidade nin do seu mecanismo, isto será obxecto da
cinética química.
 |
|
DEFINICIÓNS BÁSICAS
|
|
Sistema é unha cantidade de materia que nós illamos,
ben sexa na realidade ou imaginativamente, para estudala. Os sistemas
termodinámicos poden ser:
- Abertos, neste caso intercambian cos arredores materia e
ademais enerxía.
- Pechados, cando só intercambian enerxía. A súa masa permanece
constante, non entra nin sae materia.
- Illados, cando non intercambian cos arredores (o medio
ambiente) nin materia nin enerxía.
Todo sistema caracterízase macroscópicamente por uns
valores concretos das súas propiedades observables experimentalmente,
chamadas variables de estado, por exemplo: a presión (P), o
volume (V), a temperatura (T) e a composición (χi). Para unha substancia
pura a composición, ao ser sempre a mesma, non fai falta especificala. É
suficiente, neste caso, para coñecer o estado do sistema con tres
variables, P, V, T. Hai variables que non cambian o seu
valor aínda que dividamos o sistema, non dependen da cantidade de masa,
dise que son intensivas e outras que si dependen da cantidade de
masa, chamámoslles extensivas. Son intensivas: presión,
temperatura, densidade, composición... e son extensivas: volume, masa, e
outras como enerxía interna, entalpía, enerxía libre, entropía...
Cando se modifica unha, ou máis, variables de estado dicimos que o
sistema sufriu un proceso de cambio ou transformación.
Neste curso estudaremos unicamente sistemas que se atopan en estado de
equilibrio termodinámico. Por sorte, para describir os devanditos
sistemas só fai falta coñecer un reducido número de variables
termodinámicas. Por exemplo, se o sistema é un gas chega con coñecer a
presión, o volume e a temperatura para describir perfectamente o seu
estado. En xeral, as variables de estado atópanse relacionadas entre si
mediante ecuacións matemáticas; por exemplo: P V = n R T. (Ecuación de
estado) Existen algunhas variables termodinámicas que
teñen a importantísima calidade de que o seu valor depende
exclusivamente do estado do sistema, e non das formas intermedias polas
que este evoluciona. A estas variables denomínaselles funcións de
estado. Son funcións de estado: o volume, a enerxía interna, a
entropía, a entalpía, a presión, a temperatura, porque o seu valor, nun
momento dado, é independente do mecanismo que siga o proceso cando se
pasa dun estado inicial a outro final. As variacións que experimentan
estas funcións só dependen do estado inicial e final do sistema, sexa
cal sexa o camiño de transformación.
|
|
PRIMEIRO PRINCIPIO DA
TERMODINÁMICA
|
| Este principio reflicte a lei de
conservación da enerxía para un sistema termodinámico, e o seu obxectivo
é controlar os intercambios enerxéticos que teñen lugar entre el e a súa
contorna. |
|
O Primeiro principio da
Termodinámica establece que a enerxía dun sistema consérvase sempre.
Se ao experimentar un proceso diminúe a enerxía do sistema debe
aparecer unha cantidade equivalente de enerxía na súa contorna. |
|
Cando nun sistema pechado prodúcese un aumento ou
diminución da súa enerxía total, implicará necesariamente unha
diminución ou aumento, respectivamente, da mesma cantidade na súa
contorna.
De todas as posibles formas de enerxía que producen ou
consomen as reaccións químicas (calor, traballo, enerxía eléctrica,
enerxía luminosa etc.) neste tema unicamente vanse a considerar a
calor e o traballo. De feito, o primeiro principio da
termodinámica explica como afectan os intercambios de calor e traballo á
enerxía dun sistema; para iso emprégase unha magnitude denominada
enerxía interna que se representa por U. Con estes tres
conceptos: enerxía interna (U), traballo (W) e calor (Q) pode enunciarse
o primeiro principio da termodinámica como:
|
|
"A variación da enerxía
interna dun sistema é igual á calor desprendida ou absorbida polo
sistema máis o traballo realizado por ou sobre o sistema" |
|
Criterio de signos.
Para que o enunciado do primeiro principio sexa coherente
fai falta especificar a dirección na que flúe a enerxía, xa sexa en
forma de calor ou de traballo. Para iso adóptase un criterio de signos
que debe manterse sempre.
O criterio proposto fundaméntase nas seguintes
apreciacións:
Cando o sistema cede calor ao exterior a súa enerxía
total diminúe, a variación de enerxía interna debe ser negativa (ΔU<0) e
por tanto a calor debe ser negativo. Pero se o sistema absorbe calor da
contorna a súa enerxía total aumenta (ΔU>0), por conseguinte, esta calor
debe ser positivo.
Cando o sistema expándese realiza traballo sobre a súa
contorna á conta da súa enerxía total, que debe diminuír (ΔU<0). O
traballo de expansión debe ser negativo. Nunha compresión, a enerxía
total aumenta (ΔU>0) e o traballo de compresión debe ser positivo.
Traballo de expansión dun gas:
Para manter un gas a presión nun cilindro hai que realizar
unha forza F. Se a presión interna do gas consegue vencer a forza, o gas
expándese e realiza un traballo que vale
onde, F, é a forza que se realiza desde o exterior
sobre o émbolo para manter o gas a presión e, d, é o
desprazamento do émbolo.
A presión do gas no interior, P, é igual ao
cociente entre a forza, F, e a superficie, S, do émbolo:
se substituímos a forza da expresión do traballo obtense:
O produto da superficie do émbolo polo seu desprazamento (S·d)
é o cambio de volume que experimenta o sistema co que finalmente podemos
escribir:
que é a expresión do traballo de expansión dun gas. Lembra
que o traballo de expansión segundo o noso criterio de signos é negativo
pois se realiza á conta da enerxía interna do sistema. Nesta expresión V
é o volume final e V0 o volume inicial.
Expansión ⇒ V>V0 ⇒ − P·(V−V0)<0 ⇒
ΔU<0
Compresión ⇒ V<V0 ⇒ − P·(V −V0)>0 ⇒
ΔU>0
Comentarios sobre a enerxía interna:
A enerxía interna dun sistema, U, é a suma de todas as
enerxías contidas no devandito sistema. Considérase que a enerxía total
dun sistema gaseoso é a suma das enerxías cinéticas das súas moléculas:
U = Et + Er + Ev + Ee +
En
Pódese apreciar que hai termos debidos á enerxía
cinética das moléculas, enerxías de translación, rotación e vibración, e
termos debidos á enerxía potencial, como a atracción electrostática
entre as moléculas, e ata termos debidos ás forzas nucleares.
A enerxía interna dun sistema (U) é unha función de
estado, e o seu valor absoluto é descoñecido; o único que se pode facer
é medir a súa variación cando no sistema termodinámico se produza unha
transformación. A esta variación chamaráselle variación de enerxía
interna (ΔU).
EXERCICIOS
PARA PRACTICAR
|
|
CALORES DE REACCIÓN.
ENTALPÍA
|
| Nunha reacción química ΔU representa a
diferenza de enerxía interna entre o seu estado final (produtos) e o seu
estado inicial (reactivos), co que o primeiro principio pódese expresar:
Os valores de Q e W refírense para os efectos de calor e
traballo que acompañan a unha reacción química.
Aínda que o primeiro principio pode aplicarse a unha
reacción en calquera estado, sen limitación algunha, neste caso
referirémonos a dous casos especialmente sinxelos: ao cálculo de ΔU cando o
volume permanece constante (ΔUv) e ao cálculo de ΔU cando a
presión permanece constante (ΔUp), sendo este segundo caso o que
máis nos interese.
Transformacións a volume constante:
Neste caso, como non hai variación de volume, o traballo é
nulo, co que a expresión do primeiro principio queda:
onde Qv é o intercambio de calor a volume
constante. Esta condición dáse cando levamos a cabo a reacción nunha
bomba calorimétrica na que non pode haber contracción ou dilatación do
sistema. Nos procesos a volume constante, o valor da calor medida na
bomba calorimétrica dá directamente a variación da enerxía interna, U,
para a reacción que se está estudando.
A calor absorbida ou desprendida nunha reacción química a
volume constante (Qv) é igual á variación de enerxía interna do
sistema.
Se Q>0 ⇒ ΔU>0 ⇒ U aumenta
Se Q<0 ⇒ ΔU<0 ⇒ U diminue
Transformacións a presión constante, concepto de entalpía:
A maioría das reaccións realízanse a presión
constante, xa que nos laboratorios trabállase a miúdo en recipientes
abertos, ou o que é o mesmo, a presión atmosférica. Case todos os
procesos que imos estudar son deste tipo.
Para calcular o intercambio de calor asociada a unha
reacción a presión constante, emprégase tamén o primeiro principio. Nos
procesos a presión constante é frecuente que a medida que transcorre a
reacción exista un pequeno cambio de volume, que producirá un traballo de
expansión:
sendo ΔV = V productos – V reactivos, co que
o primeiro principio toma a forma:
onde Qp = variación de calor a presión constante.
Despexando o calor Qp:
Desenvolvendo esta expresión:
e se se agrupan os termos correspondentes aos
estados inicial e final obtense:
P·V ten dimensións de enerxía e sumada á enerxía interna U
daranos unha nova enerxía que chamaremos entalpía e que
representaremos por H:
que substituída na ecuación anterior dános a calor
intercambiada nunha reacción a presión constante:
A entalpía, H, é outra forma de medir a enerxía dun
sistema; H e H0 son os valores da enerxía calorífica do
sistema nos estados final e inicial, e a súa diferenza, ΔH,
representa a calor transferida polo sistema en calquera proceso que teña
lugar a presión constante.
La entalpía es una función de estado, e igual que le ocurría a la energía interna, no se pode medir su valor absoluto, sólo se
puede medir su variación (en procesos a presión constante).
A entalpía é unha función de estado, e igual que lle ocorría
á enerxía interna, non se pode medir o seu valor absoluto, só se pode medir
a súa variación (en procesos a presión constante).
Se H>H0 ⇒ ΔH>0 ⇒ Qp>0
o sistema absorbe calor, logo o proceso é endotérmico.
Se H<H0 ⇒ ΔH<0 ⇒ Qp<0
o sistema desprende calor, logo o proceso é exotérmico.
|
|
ENTALPÍA DE REACCIÓN.
DIAGRAMAS ENTÁLPICOS
|
| Como a entalpía é unha función de estado,
cando un sistema evoluciona desde un estado inicial a outro final, a súa
variación de entalpía exprésase como:
No caso dunha reacción química o estado inicial son os
reactivos e o estado final son os produtos, por tanto a variación de
entalpía dunha reacción será:
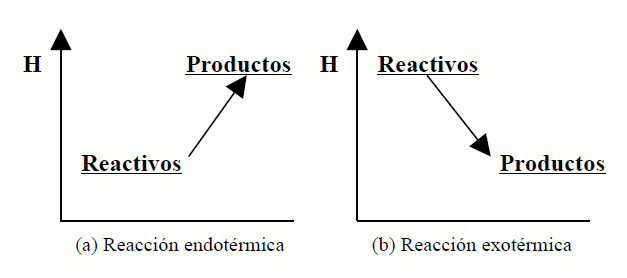
Se Hp>Hr ⇒ ΔH>0
⇒ Qp>0, (o sistema absorbe calor) a reacción é endotérmica.
Se Hp<Hr ⇒ ΔH<0 ⇒
Qp<0, (o sistema desprende calor) a reacción é exotérmica.
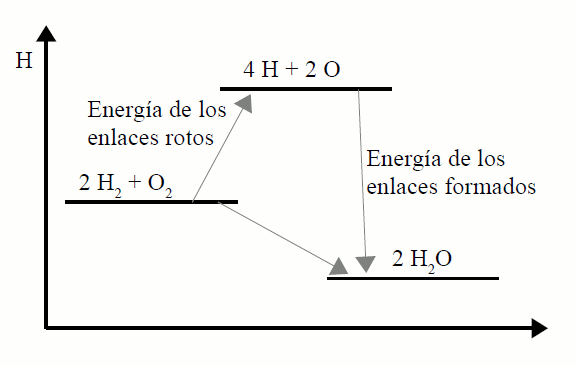
A variación de entalpía dunha reacción pódese expresar
gráficamente con axuda dos diagramas entálpicos, nos que se representa
sobre unha escala de enerxías a entalpía de reactivos e produtos. Ao non
poderse coñecer o valor absoluto, a escala de entalpías (en ordenadas)
ten unha orixe arbitraria. A figura (a) representa o diagrama entálpico
para unha reacción endotérmica, e a (b) para unha reacción exotérmica.
Ecuaciones termoquímicas:
En termoquímica, para representar unha reacción, ademais
de indicar os coeficientes estequiométricos indícase o estado (sólido,
líquido ou gas) de cada unha das substancias. Tamén debe incluírse o
intercambio de calor (ganancia ou perda) que ten lugar no proceso. Esta
forma de representar a ecuación recibe o nome de ecuación
termoquímica. A inclusión da calor intercambiada no proceso pódese
facer de dúas maneiras: poñendo explicitamente a calor, ou poñendo o
valor que para ese proceso ten ΔH ou ΔU. Neste último caso podemos
saber, ademais, se o proceso ocorre a presión ou a volume constante.
Exemplo:
|
|
MEDIDAS CALORIMÉTRICAS
|
As reaccións químicas poden realizarse dentro
dun recipiente hermeticamente pechado, indeformable mecanicamente, de
paredes ríxidas que impidan calquera variación de volume do sistema no
transcurso das mesmas. Se as paredes do recipiente permiten o paso libre de
calor a través do mesmo, a cantidade de calor intercambiada cando ten lugar
a reacción química chámase calor de reacción a volume constante.
Estas calores de reacción a volume constante mídense
mediante dispositivos como o da figura chamados bomba calorimétrica.
Para medir unha calor de combustión a mostra pesada introdúcese nun
crisol (que se introduce á súa vez na bomba) e quéimase completamente en
osíxeno a presión. A mostra quéntase mediante un filamento de ignición
de ferro, que se pon incandescente cando se pasa unha corrente eléctrica
procedente dunha batería. O calorímetro está illado mediante un manto
illante térmico. A temperatura do fluído calorimétrico mídese co
termómetro. A partir do cambio de temperatura e tendo en conta a
cantidade de calor engadida a través do filamento, pódese calcular a
calor de reacción.
Este mesmo proceso pódese realizar nun recipiente dotado
dunha parede móbil, sobre a que actúa unha presión constante (en xeral a
atmosférica). Despois de restablecida a temperatura inicial, terminada a
reacción, ás veces obsérvase que o volume inicial non é igual ao volume
final, pois o número de moles de gas dos reactivos non ten que coincidir co
número de moles de gas dos produtos. Supoñendo comportamento ideal dos gases
que participan na reacción podemos establecer que:
Como podemos facer medidas calorimétricas nun
calorímetro?
Primeiro lembraremos como medir a calor:
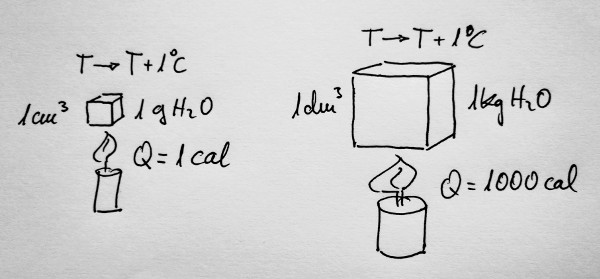
Fagamos un experimento: imos achegar calor a unha certa
cantidade de auga.
Se achegamos calor a un gramo de auga para que a súa
temperatura aumente un grao centígrado, ou un grao Kelvin, a esa
cantidade de calor chamóuselle caloría. E represéntase por cal.
Da mesma forma, se achegamos calor a un quilogramo de auga
para que a súa temperatura aumente un grao centígrado, ou un grao Kelvin,
esa cantidade de calor serán 1000 calorías.
Pero a caloría non é a unidade de enerxía no Sistema
Internacional de unidades, esa unidade é o Julio. A equivalencia entre
xullos e calorías é:
Por tanto, a calor que temos que achegar a 1kg de auga
para que a súa temperatura aumente 1ºC, ou 1K, é 4180J. Esta cantidade
coñécese como a calor específica da auga.
É un valor que podemos determinar para cada substancia
pura e é de gran utilidade para medir calores.
Calor específica dunha substancia, c, é a calor que hai
que proporcionar a unha unidade de masa, 1kg, para que a súa temperatura
aumente 1K. A súa unidade é J/kg K. Por exemplo, a calor
específica da auga é 4180J/kg K, xa que hai que achegar 4180J a un
quilogramo de auga para que a súa temperatura aumente un Kelvin, ou un grao
centígrado.
|
Substancia |
Calor
específica, c (J/kg K) |
Substancia |
Calor
específica, c (J/kg K) |
| Auga (liq.) |
4180 |
Ouro |
129 |
| Xeo |
2114 |
Prata |
237 |
| Vapor de auga |
2080 |
Mercurio |
139 |
| Etanol |
2440 |
Cobre |
385 |
| Amoníaco (liq.) |
4700 |
Hidróxeno |
14300 |
| Aluminio |
897 |
Nitróxeno |
1040 |
| C (grafito) |
710 |
Oxíxeno |
918 |
| Ferro |
450 |
Area |
835 |
| Chumbo |
129 |
Granito |
790 |
A calor absorbida ou cedida por un corpo calcúlase coa
seguinte ecuación:
Onde (Q) é a calor cedida ou absorbida, (m) a masa, (c) a
calor específica , (T) a temperatura final, e (To) a temperatura
inicial
Un calorímetro é un aparello que nos vai a permitir
calcular calores de reacción, que por ser a presión constante serán
entalpías. A súa construción debe garantir que a calor non saia do mesmo,
aínda que o mesmo poida absorber certa cantidade de calor.
Nun calorímetro:
Pois consérvase a enerxía.
A calor cedida ou absorbida por unha substancia podémola
calcular a partir de
onde m é a masa, c a calor específica e ΔT a variación da
temperatura.
O calorímetro tamén absorberá unha certa cantidade de
calor que debemos calcular para cada calorímetro.
onde Ccal é a capacidade calorífica do
calorímetro. Como determinar a capacidade calorífica
do calorímetro?
Introducimos unha certa cantidade de auga no calorímetro a
temperatura ambiente, o calorímetro tamén estará a esta temperatura, que
chamaremos Tf. Quentamos outra cantidade de auga e medimos a
súa temperatura, que chamaremos Tc. Introducimos rapidamente
esta cantidade de auga no calorímetro. Medimos a temperatura que
chamaremos Teq. A calor cedida pola auga quente debe
coincidir coa calor absorbida pola auga fría e o calorímetro.
Este valor conservarémolo para todas as experiencias con ese calorímetro.
Como determinar un calor de disolución?
Engadimos unha cantidade de auga ao calorímetro e medimos
a temperatura. Logo medimos unha masa de sal e engadímola ao
calorímetro. Axitamos ata a disolución completa. Medimos a temperatura
de equilibrio e realizamos os cálculos:
A calor específica da disolución podemos asimilala á calor
específica da auga sen cometer moito erro para disolucións diluídas.
Práctica de calorimetría: Medida de calores de
reacción, e comprobación da lei de Hess:
EXERCICIOS
PARA PRACTICAR
|
|
ENTALPÍA DE REACCIÓN
|
| A variación de entalpía que se produce durante
unha reacción a presión constante pódese expresar como a diferenza entre as
entalpías dos produtos e as entalpías dos reactivos.
Pero non podemos coñecer as enerxías absolutas asociadas a
cada substancia. Xa o vimos tamén para o caso da enerxía interna. Como
calcular entón as entalpías das reaccións? Temos catro formas de facelo:
- Mediante medidas calorimétricas.
- Mediante a lei de Hess.
- Mediante entalpías de formación.
- Mediante entalpías de enlace.
|
|
LEI DE HESS
|
| A entalpía é unha función de estado e a súa
variación ΔH a presión constante só depende dos estados inicial e final,
pero non dos intermedios polos que transcorre a reacción.
Baseándose nesta propiedade, o suízo Germain Henri Hess formulou en 1840
o principio que leva o seu nome:
|
|
Lei de Hess: Cando unha reacción
química pode expresarse como suma alxebraica doutras, a súa calor de
reacción é igual á suma algebraica das calores das reaccións
parciais. |
|
En moitos casos non é posible determinar experimentalmente a
entalpía de reacción dun proceso; por exemplo a entalpía de formación do
CO, non é medible nun calorímetro. Nestes casos a lei de Hess é
especialmente útil:
A partir destas dúas reaccións podemos calcular a calor da
primeira. Teremos que colocalas de forma que sumadas déannos a que
buscamos, o mesmo que facemos coas ecuacións facémolo coas calores. Como
colocalas? Buscamos en cada ecuación a substancia clave que é a
substancia da ecuación problema que aparece en exclusiva en cada
ecuación, esa substancia colocámola na mesma posición e co mesmo
coeficiente que na ecuación problema:
A ecuación A deixámola tal cal, pois o carbono aparece
como reactivo e con coeficiente 1. Pero a B invertímola pois o CO
aparece como reactivo e na ecuación problema como produto aínda que co
mesmo coeficiente. O mesmo que facemos coas ecuacións facémolo coas
calores.
Sumamos e comprobamos que nos dá a ecuación problema, o mesmo facemos
coas calores.
EXERCICIOS
PARA PRACTICAR
|
|
ENTALPÍA NORMAL DE
FORMACIÓN
|
|
Outra forma de poder calcular a entalpía dunha
reacción podería ser tabulando os valores da entalpía de calquera reacción.
Pero como podes imaxinar isto é pouco menos que misión imposible. Se se
tivese que tabular a ΔH para todas e cada unha das miles de reaccións que se
poden realizar a presión constante, seguramente non atopariamos sitio para
almacenar tanta información.
Un composto determinado pode intervir en miles
de reaccións químicas pero só se tabula unha delas; aquela na que se forma
este composto a partir dos seus elementos no estado de agregación (sólido,
líquido ou gas) no que se atopan nas condicións habituais de laboratorio
(condicións estándar): presión de 1atm e temperatura de 298K (25ºC).
Ás entalpías dos elementos, no estado de
agregación máis estable nas condicións anteriores, asígnaselles o valor cero
pola propia definición de entalpía de formación.
A entalpía normal de formación dun composto
en condicións estándar, tamén denominada calor de formación, representarase
por ΔHºf, e defínese como: o cambio de entalpía, ΔH, que ten
lugar cando se forma un mol de composto a partir dos elementos que o
constitúen nos seus estados de agregación máis estables en condicións
estándar.
A entalpía é unha forma de enerxía, por tanto é
unha magnitude extensiva que depende da masa do sistema; por esta razón, ao
definir a entalpía de formación especifícase que se refire á formación dun
mol de composto. Exemplos:
Ag(s) + 1/2 Cl2(g) → AgCl(s) ΔH = −
127 kJ/mol
1/2N2(g) + 1/2 O2(g) → NO(g) ΔH = 90,4 kJ/mol
1/2 N2(g) + 3/2 H2(g) → NH3(g) ΔH = −
46,0 kJ/mol
Se observas a maioría das calores de formación en
condicións estándar nunha táboa comprobarás que son negativos, polo que
devanditos procesos serán exotérmicos, e as descomposicións dos
compostos serán, por conseguinte, endotérmicas.
TÁBOA
DE DATOS QUÍMICOS. Nesta táboa podes atopar tabuladas as
entalpías de formación de moitas substancias.
|
|
ENTALPÍA DE REACCIÓN A
PARTIR DE ENTALPÍAS DE
FORMACIÓN
|
| Como se indicou anteriormente de forma
xenérica a variación da entalpía dunha reacción é:
Agora estamos capacitados para realizar o cálculo da
entalpía dunha reacción en condicións estándar de forma exacta, xa que
aínda que non coñecemos as entalpías absolutas se podemos asociar a cada
substancia a súa entalpía de formación nas condicións estándar.
As entalpías de formación nas devanditas condicións
corresponden á formación dun mol de substancia; por iso, ao ser a entalpía
unha magnitude extensiva, cando na ecuación axustada os coeficientes das
substancias que interveñen na reacción sexan diferentes de 1, será preciso
multiplicar as entalpías de formación polos coeficientes estequiométricos
para calcular a entalpía da reacción. A entalpía de reacción en condicións
estándar pódese escribir como:
onde: np e nr son os coeficientes
estequiométricos dos produtos e dos reactivos.
Esta ecuación é unha aplicación de que a entalpía é unha función de
estado, e por tanto da lei de Hess.
EXEMPLO 1: Calcula a entalpía normal para a reacción de combustión do etanol
C2H5OH(l)
Axustamos a reacción: C2H5OH(l) + 3
O2(g) → 2 CO2(g) + 3 H2O(l)
Dado que: ΔHºR = Σ np·ΔHºfp − Σ
nr·ΔHºfr
e que ΔHºf [O2(g)] = 0
utilizando a
táboa
de datos químicos.
ΔHºR = 2mol·ΔHºf [CO2(g)] + 3mol·ΔHºf
[H2O(l)] − 1mol·ΔHºf [C2H5OH(l)] =
= 2mol (− 393,7kJ/mol) + 3mol (− 285kJ/mol) − 1mol (− 277kJ/mol) = − 1365,4 kJ
EXERCICIOS
PARA PRACTICAR
|
|
AS ENERXÍAS DOS ENLACES
E A SÚA RELACIÓN COA CALOR DE REACCIÓN
|
|
Ás veces non se coñecen as entalpías de formación de
determinados compostos. Un método aproximado para calcular calores de
reacción, sabendo que nunha reacción química rompen uns enlaces e fórmanse
outros novos, é asignar a cada enlace a enerxía que necesitamos para
rompelos.
A enerxía de enlace ou entalpía de enlace defínese como: o
fluxo de enerxía cando rompe un mol de enlaces en estado gaseoso a presión
constante.
A entalpía de enlace sempre é positiva, xa que é a enerxía
que absorbe a molécula cando rompe un dos seus enlaces químicos.
O concepto de enerxía de enlace axúdanos a entender por que
algunhas reaccións son exotérmicas e outras endotérmicas, pois se os enlaces
das moléculas dos produtos son máis fortes que os enlaces das moléculas
reaccionantes, a reacción será exotérmica, xa que:
No caso de que se pase de enlaces débiles nos reactivos a
enlaces máis fortes nos produtos, producirase un desprendemento de
enerxía e ΔH será negativa, polo que a reacción será exotérmica.
Os valores das entalpías de enlace son valores medios
calculados a partir de diferentes compostos. Por tanto serán valores
aproximados, aínda que sempre será mellor ter un valor aproximado dunha
magnitude que non ter ningún.
O cálculo das entalpías de reacción a partir das entalpías
de enlace é especialmente interesante nos compostos orgánicos, debido ao
inmenso número destes que fai que non se dispoña das entalpías de formación
de moitos compostos, ademais de posuír moi poucos tipos de enlace, así que
cunha táboa pequena de entalpías de enlace tabuladas podemos calcular
moitísimas entalpías de reacción. Para o cálculo será fundamental coñecer os
enlaces que se dan nos compostos, para o que axuda coñecer as fórmulas
desenvolvidas dos compostos que forman a reacción.
EXEMPLO 2: Calcula a ΔHcombustión a 298K do propano CH3‒CH2‒CH3 a partir dos seguintes datos: ΔHº(C‒C)=348, ΔHº(C‒H)=413, ΔHº(O‒H)=463, ΔHº(O=O)=495, ΔHº(C=O)=802 todos os valores en kJ/mol
ΔHºR = 8mol · ΔHº(C‒H) + 2mol · ΔHº(C‒C) + 5mol
· ΔHº(O=O) ‒ 6mol · ΔHº(C=O) ‒ 8mol · ΔHº(O‒H) =
= 8mol · 413kJ/mol + 2mol · 348kJ/mol + 5mol · 495kJ/mol ‒ 6mol ·
802kJ/mol ‒ 8mol · 463kJ/mol = ‒2041kJ
Cando utilizamos entalpías de formación o resultado é de
‒2044,5 kJ, é un resultado aproximado, pero será de gran utilidade cando non
teñamos datos de entalpías de formación, ou non podamos calcular
experimentalmente dita entalpía.
EXERCICIOS
PARA PRACTICAR
|
|
ESPONTANEIDADE.
CONCEPTO DE ENTROPÍA |
|
Tendo en conta o estudado anteriormente podemos
chegar á seguinte conclusión: dende o punto de vista enerxético, existen
dous tipos de reaccións principais:
a) As que desprenden calor ao formarse os
produtos a partir dos reactivos mediante unha transformación química, é
dicir, son exotérmicas e, por tanto, energéticamente favorables.
b) Aquelas nas que necesitamos unha achega
continua do exterior para que se poidan realizar, pois nunca se alcanza un
mínimo enerxético, e en principio non serán favorables dende este punto de
vista. Son endotérmicas.
Agora ben, parece que o factor enerxético non é
o único que intervén na espontaneidade dunha reacción, pois existen na
natureza procesos energéticamente desfavorables (endotérmicos), que teñen
lugar dunha maneira espontánea, por exemplo a fusión do xeo e a evaporación
da auga. De igual maneira, a disolución do sal común en auga é un proceso
endotérmico que transcorre espontaneamente:
NaCl(s) + H2O(l) → Cl–(aq) +
Na+(aq) ; ΔH = 3,9 kJ
Tamén se descompón espontaneamente o
carbonato de amonio en hidroxenocarbonato de amonio e amoníaco:
(NH4)2CO3 → (NH4)HCO3 +
NH3 ; ΔH = 40 kJ
Por tanto, o factor enerxético non será o
único que deba terse en conta cando se trate de predicir se unha
reacción química vai ter lugar de maneira espontánea ou non.
Para comprender algunhas das condicións que
determinan se un proceso pode ou non realizarse, é útil observar algunhas
transformacións que se realizan espontaneamente sen achega enerxética
exterior. Exemplos destas transformacións son:
-
a expansión dun gas nun espazo baleiro
-
a difusión de dous gases dentro dun recipiente ata que a
mestura sexa homoxénea
-
a disolución dun sólido en auga
-
a condutividade de calor ao longo dunha barra de metal
que ten un extremo quente e o outro frío
Unha característica fundamental destes procesos
é que non se invirten por si sós. Para que se invirtan requírese a acción
dun axente externo, é dicir, os procesos espontáneos non son
termodinámicamente reversibles (son procesos irreversibles); o
coñecemento deste feito experimental foi a base do segundo principio da
Termodinámica.
A historia da segunda lei da Termodinámica
empeza co francés Sadi Carnot. Carnot estaba interesado na eficiencia
dos motores térmicos. O seu motor extraía calor dun foco quente e cedía
calor a un foco frío, producindo una certa cantidade de traballo. A
eficiencia do motor ideal de Carnot non podía ser igualada ou superada por
ningún motor real.
Estas ideas de Carnot foron a base para que o
inglés William Thomson e o alemán Rudolf Clausius formulasen o
Segundo principio da Termodinámica.
A primeira formulación do segundo principio
debémoslla a Kelvin.
|
|
Non pode haber ningún
motor cíclico cuxo único efecto sexa extraer enerxía dun reservorio
de calor e convertela completamente en traballo. |
|
Por tanto establece que, aínda que a calor é
unha forma de enerxía, é imposible converter completamente calor (ou enerxía
térmica) en traballo. Aínda que o contrario si é posible, pódese converter
todo o traballo en calor.
A formulación de Clausius é unha evidencia que
temos todos,
|
|
A calor sempre flúe dun
corpo máis quente (a máis temperatura) a un corpo máis frío (a menos
temperatura). |
|
Nunca se observa o proceso inverso de forma
espontánea. Da mesma forma que a calor sempre flúe nun sentido xa vimos
antes que outros procesos irreversibles tamén tiñan lugar sempre nun
determinado sentido. Pero por que? É Clausius quen propón que hai unha
magnitude que rexe estes procesos e que chamou entropía, S. A
entropía, é función de estado e extensiva. Pero que mide realmente a
entropía?
Procesos irreversibles: Na natureza hai
procesos que partindo dunha situación inicial transcorren nun sentido, sen
que se poida recuperar a situación inicial, a non ser que interveña un
axente externo. Se se interconectan dous recipientes que conteñen gases
diferentes, ao transcorrer o tempo e sen necesidade de ningunha acción
exterior, os gases combínanse ata converterse nunha mestura homoxénea. Pola
contra, dous gases que formen unha mestura homoxénea nunca se van a separar,
por moito tempo que se deixe evolucionar o sistema.
Procesos reversibles: Outros procesos
transcorren a través dunha sucesión de estados de equilibrio, de xeito que
en calquera instante póidase invertir o proceso e facelo evolucionar en
sentido contrario ata o estado inicial.
Segundo Clausius o segundo principio da
Termodinámica podémolo formular da seguinte maneira:
|
|
O Segundo principio da
Termodinámica: "Nun proceso reversible a entropía é constante pero,
se é irreversible, a entropía aumenta" |
|
En física estabamos afeitos a que nos
procesos de cambio rexían leis de conservación, conservación da masa, da
enerxía, do momento, da carga, pero unha magnitude que aumenta sempre
non é fácil de entender.
Universo mecánico 47: Entropía
Para entender de forma cualitativa o concepto
de entropía acódese ás veces ao concepto de orde e desorde. Esta non é a
mellor forma de achegarnos ao concepto de entropía pero como primeira
aproximación pode ser válida.
Que entendemos por orde e desorde? O que
pode ser orde para unha persoa pode non selo para outra. Dise ás veces
que os corpos moi desordenados teñen unha elevada entropía; pola contra,
os corpos ou sistemas moi ordenados posúen baixo contido entrópico. Isto
fai que, cando un sólido funde, se evapora ou se disolve, as súas
moléculas aumentan a capacidade de moverse (aumentan os graos de
liberdade), diremos que están máis desordenadas, ou o que é equivalente,
a súa entropía aumenta. Tentaremos aclarar máis adiante estes conceptos.
Nun sistema illado, que non pode
intercambiar coa contorna nin materia nin enerxía, é posible un cambio
se iso supón un aumento de entropía; con todo, a evolución do sistema
detense cando a entropía é máxima.
Un sistema pechado, que non intercambia
materia coa contorna, pero si enerxía, pode evolucionar de dúas maneiras:
a) Mediante un proceso reversible no
cal a entropía gañada ou perdida polo sistema debe coincidir coa perdida
ou gañada pola contorna:
ΔSsistema = – ΔSentorno
ΔStotal = 0 (Proceso isoentrópico)
b) Mediante un proceso irreversible no
cal se inclúen a maioría dos procesos que transcorren espontaneamente,
xa que o sistema evoluciona cara a unha maior desorde, o que supón un
aumento de entropía, ΔS>0.
Ambos casos podémolos resumir coa
expresión:
Cuantitativamente, a variación de entropía
dun sistema que pasa dun estado inicial a un estado final mediante un
proceso reversible defínese por:
E se o proceso é irreversible:
onde en ambos casos ΔQ é cantidade de calor
intercambiada polo sistema, ΔS é a variación de entropía entre os
estados inicial e final, e T é a temperatura absoluta á que se produce a
transformación.
Unha mellor comprensión do concepto de
entropía debémoslla ao físico austríaco Boltzmann.
Boltzmann acertou de pleno ao propoñer unha
interpretación estatística da entropía. Nun momento no que non estaba
asentada a teoría atomista da materia as súas ideas non foron ben
aceptadas cando non rexeitadas. Quen sabe se foi este rexeitamento ás
súas ideas o que o levou ao suicidio en 1906. As súas achegas foron
fundamentais para a teoría cinética dos gases e da calor.
Aínda que a termodinámica non fai
suposicións acerca da estrutura da materia, pode mellorarse a nosa
comprensión das funcións termodinámicas se tratamos de interpretalas en
termos das propiedades moleculares. Sabemos que a presión dun gas
resulta das colisións moleculares coas paredes do recipiente, que a
temperatura é un parámetro que expresa a enerxía cinética media das
moléculas, e que a enerxía interna consiste nas enerxías cinética e
potencial de todos os átomos, moléculas, electróns e núcleos dun
sistema. Que propiedade molecular reflicte a entropía?
Existen dúas maneiras de describir o estado
dun sistema termodinámico: a descrición macroscópica dada polos valores
das funcións de estado, tales como P, V e T, e a descrición microscópica
que implicaría dar a posición e velocidade de cada átomo do sistema.
Pensemos para un mol a cantidade tan inmensa de información que
necesitariamos, e isto só para un instante.
Cando observamos calquera sistema
termodinámico nun estado de equilibrio macroscópico, o seu estado
microscópico está a cambiar a unha velocidade enorme. A pesar desta
actividade molecular, as propiedades dun estado macroscópico permanecen
constantes. Isto debe significar que existen moitos estados
microscópicos compatibles con calquera estado macroscópico. A
entropía é unha medida do número de estados microscópicos asociados cun
estado macroscópico particular.
Para explorar este punto utilicemos unha
baralla de cartas como un símil dun sistema termodinámico. Existen dous
estados macroscópicos distintos da baralla: ou está "ordenada", coas
cartas nalgunha secuencia normal; ou está "desordenada", coas cartas
nunha secuencia ao chou. Podemos ver que só hai un estado microscópico
"ordenado". Pero existen moitos estados microscópicos asociados co
estado macroscópico "desordenado", porque hai moitas secuencias ao chou
para as cartas. Posto que a entropía mide o número de estados
microscópicos do sistema, e aumenta co devandito número, podemos dicir
que o estado "desordenado" ten unha entropía máis alta que o estado
"ordenado". Se se nos caen as cartas ao chan e recollémolas rapidamente,
o máis probable é que as cartas estean desordenadas, pois hai máis
combinacións nas que as cartas estean desordenadas que combinacións en
que as podamos considerar ordenadas. Para as cartas un estado ordenado é
menos probable que un estado desordenado.
Empregando esta análise podemos ver por que
unha baralla de cartas cambia dun estado macroscópico "ordenado" a un
estado "desordenado" cando as cartas son baralladas. Debido a que hai
máis estados microscópicos asociados co estado macroscópico
"desordenado", para a baralla é simplemente máis probable ir parar a
unha situación de maior "desorde". Se aplicamos este razoamento á
conduta dos sistemas termodinámicos, podemos ver que a entropía ten unha
tendencia natural cara ao aumento, dado que isto corresponde ao cambio
dos sistemas desde as condicións de baixa probabilidade aos estados de
maior probabilidade. É cuestión de estatística.
Por que estes estados "desordenados" son
máis probables que os estados "ordenados"? A resposta está no que
realmente entendemos por "desorde". Un sistema desordenado é aquel de o
cal temos unha cantidade relativamente pequena de información referente
ao seu estado microscópico exacto. A razón pola cal carecemos deste
coñecemento detallado é que o sistema ten moitos estados microscópicos
asociados a el, e o mellor que podemos facer é supoñer que en calquera
instante el está nalgún dos mesmos. Se soamente fosen posibles uns
cuantos estados microscópicos, poderiamos ser capaces de facer unha
conxectura exacta daquel no que estaba o sistema e, sendo así, facer
unha descrición detallada das posicións e velocidades das moléculas.
Deste xeito, un sistema "desordenado" é aquel que ten un número
relativamente grande de estados microscópicos asociables a el, e isto é
o porqué de que un estado desordenado sexa máis probable que un estado
ordenado. Este vídeo pódeche axudar a entendelo.
Cando falamos da entalpía atopamos útil
seleccionar a un certo estado da materia e asignarlle unha entalpía de
formación definida. A nosa decisión de que a entalpía de formación de
todos os elementos nos seus estados estándar sexa cero baséase soamente
na conveniencia. A calquera outro estado dos elementos poderíaselle
asignar a entalpía cero. No caso da entropía o caso é diferente, porque
a asociación da entropía co número de estados microscópicos dispoñibles
para un sistema suxire unha selección natural da entropía cero. Nun
cristal perfecto, no cero absoluto, hai un só estado microscópico
posible. Cada átomo debe estar nun punto da rede cristalina e debe ter
unha enerxía mínima. É así como podemos dicir que este é un estado de
orde perfecta, ou de entropía cero. Esta importante decisión é expresada
por o
|
|
Terceiro principio da
Termodinámica: "A entropía dos cristais perfectos de todos os
elementos e compostos puros é cero á temperatura do cero absoluto". |
|
A terceira lei permítenos asignar unha
entropía absoluta a cada elemento e a cada composto, a calquera
temperatura. Para un mol de substancia:
Se avaliamos esta integral desde 0K ata 298K , para un mol
de substancia a unha atmosfera de presión obtemos a entropía estándar
absoluta, Sº, que definimos como o valor da entropía molar dunha
substancia á temperatura de 25ºC e presión dunha atmosfera. Os seus
valores atopámolos tabulados, para as diferentes substancias, en J/molK.
Táboa
de datos químicos
|
|
ENTROPÍA DE REACCIÓN A
PARTIR DE ENTROPÍAS ABSOLUTAS
|
| A entropía é unha función de estado, por
tanto, a variación de entropía nunha reacción química a temperatura
constante pódese definir como:
Se expresamos esta ecuación en función das condicións
estándar, 25ºC e 1 atm de presión:
A entropía estándar de reacción, de maneira semellante a
como fixemos coas entalpías, podémola calcular a partir dos valores
tabulados das entropías absolutas.
EXEMPLO 3: Calcula a variación de entropía na reacción:
CaCO3(s) → CaO(s) + CO2(g)
Dado que: ΔSºR = Σ np·Sºp − Σ
nr·Sºr
Utilizando a
táboa
de datos químicos.
ΔSºR = 1mol·Sº [CaO(s)] + 1mol·Sº [CO2(g)]
–
1mol·Sº [CaCO3] =
= 1mol (39,7JK–1mol–1) + 1mol (213,8JK–1mol–1)
– 1mol(92,9JK–1mol–1) =
160,6 JK–1
O valor positivo que se obtén para a entropía neste exercicio pódese
predicir analizando o concepto de entropía. Para avaliar o signo da
entropía dun proceso débese ter en conta que:
- Unha reacción que orixina un aumento no número de moles de gas vai
sempre acompañada dun aumento de entropía. Se o número de moles de gas
diminúe, ΔS será negativa.
- Se nunha reacción non interveñen especies gasosas, pero existe un
aumento considerable no número de moles dos produtos respecto aos
reactivos, tamén cabe esperar unha ΔS positiva, tal é o caso das
substancias hidratadas:
CuSO4·5H2O(s) →
CuSO4(s) + 5H2O(l) ΔS>0
pois se pasa dun mol nos reactivos a seis moles nos produtos.
- Se o cambio no número de moles non é moi significativo, podemos
atopar ΔS positivas e ΔS negativas, por exemplo:
2AgI(s) → 2Ag(s) + I2(s) ΔS = – 6,2
PbI2(s) → Pb(s) + I2(s) ΔS = 1,1
|
|
CRITERIO
DE ESPONTANEIDADE. ENERXÍA LIBRE DE GIBBS
|
|
Unha vez introducido o concepto de entropía estamos en
condicións de non confundir a tendencia á realización dun proceso químico,
coa calor intercambiada no mesmo, xa que ademais deste parámetro existe
outro tan importante como o grao de desorde do sistema ou entropía.
Existe unha magnitude termodinámica que engloba e relaciona
ambos os parámetros, dunha parte a calor intercambiada no proceso, ΔH, e
doutra a desorde alcanzada no mesmo, ΔS.
Esta magnitude, ΔG, chámase enerxía libre de
Gibbs ou entalpía libre e é, como ΔH e ΔS, unha magnitude
termodinámica extensiva e función de estado:
Empregando a enerxía libre de Gibbs é moi sinxelo predicir
a espontaneidade dos procesos. Un proceso será espontáneo cando ΔG
sexa negativo, é dicir:
EXEMPLO 4: Calcula a variación de enerxía libre na reacción
seguinte en condicións estándar:
CaCO3(s) → CaO(s) + CO2(g)
Calculando a ΔHºR e a ΔSºR a partir dos datos tabulados obtemos:
ΔHºR = +177,7kJ/mol
ΔSºR = +160,6J/mol K
Utilizando a ecuación de Gibbs:
ΔGºR = ΔHºR –T·ΔSºR = 177,7kJ – 298K · 0,1606kJ/K = +129,8 kJ
Deducimos deste valor da enerxía libre, ΔG>0, que a
reacción non é espontánea á temperatura de 25ºC.
Toda reacción que transcorra con diminución de entalpía
(ΔH<0) e aumento de entropía (ΔS>0) será espontánea e a variación de enerxía
libre, ΔG, sempre será negativa. De igual maneira, aquelas reaccións nas que
ΔH sexa positivo (endotérmicas) e ΔS sexa negativo nunca serán espontáneas,
pois ΔG será sempre positivo.
Agora ben, existen reaccións onde os termos entálpico e
entrópico están enfrontados, e será a temperatura a magnitude que determine
a espontaneidade ou non do proceso. Observemos isto nun caso concreto, a
descomposición do carbonato de calcio:
CaCO3(s) → CaO(s) + CO2(g) ;
ΔH >0
1. ΔH é positivo, o que indica que a reacción é endotérmica.
2. Se pasa dunha molécula en estado sólido a unha molécula de sólido e outra de gas, co que
o desorde aumentará,
ΔS positivo.
Como tanto ΔH como TΔS son positivos, o signo da enerxía libre
ΔG = ΔH - TΔS dependerá do valor de T:
a) Se T é pequeno, ΔH será >TΔS, co que ΔG será positivo
e o proceso nunca será espontáneo.
b) Se T é grande, ΔH será <TΔS, co que ΔG será negativo
e o proceso será espontáneo.
c) Existirá un valor de T no que se cumpra que ΔH = TΔS, co que
ΔG = 0 e o proceso se atopará en equilibrio.
No seguinte cadro preséntanse as diferentes alternativas:
| ΔH |
ΔS |
ΔG |
Observacións |
| Negativa |
Positiva |
Negativa |
Espontánea a cualquera T, T non influe. |
| Positiva |
Negativa |
Positiva |
Non espontánea a cualquera T. Ocorre
o proceso inverso. |
| Positiva |
Positiva |
a T baixa
Positiva |
A T baixa non é espontánea, se ΔH>TΔS |
a T alta
Negativa |
A T alta a reacción é espontánea, se TΔS>ΔH. |
| Negativa |
Negativa |
a T baixa
Negativa |
A T baixa é espontánea, se |ΔH|>|TΔS| |
a T alta
Positiva |
A T alta non é espontánea, se |TΔS|>|ΔH| |
Lembremos por tanto que:
Se ΔG < 0 o proceso ten lugar espontaneamente, evoluciona pasando de
reactivos a produtos.
Se ΔG > 0 o proceso non é espontáneo, e ten lugar en sentido contrario
pasando de produtos a reactivos.
Se ΔG = 0 o proceso permanece en equilibrio coexistindo reactivos e
produtos.
EXEMPLO 5: Calcula a temperatura á cal esta reacción será espontánea:
CaCO3(s) → CaO(s) + CO2(g)
Sabendo que ΔHºR = +177,7kJ/mol e ΔSºR = +160,6J/mol K en condicións estándar,
e supoñendo que estos valores non varían apreciablemente coa temperatura.
Se estos valores non varían apreciablemente coa temperatura podemos calcular a que temperatura a reacción estará en equilibrio,
ΔG = 0,
Como o termo da entalpía é positivo e o da entropía
negativo, a temperaturas máis altas de 833,5ºC o termo da entropía será
maior que o termo da entalpía e así a variación de enerxía libre será
negativa, e por tanto a reacción será espontánea.
|
|
ENERXÍA
LIBRE NORMAL DE
FORMACIÓN
|
|
O mesmo que pasaba coas entalpías pasa coas enerxías libres,
non podemos coñecer os seus valores absolutos. Pero podemos tabular os
valores das enerxías libres para as reaccións de formación de cada unha das
substancias que participan nunha reacción química. Como a enerxía libre é
unha función de estado poderemos calcular a enerxía libre dunha reacción a
partir das enerxías libres de formación.
Definiremos a enerxía libre normal de formación dun composto
nas condicións estándar (T = 298K e P = 1atm) como o cambio de enerxía libre
necesario para formar un mol de composto a partir dos elementos en estado
fundamental nesas condicións de presión e temperatura. Estes valores de ΔGºf
témolos tabulados para as principais substancias. Do mesmo xeito que pasaba
coas entalpías as ΔGºf dos elementos será cero por definición de
enerxía libre de formación.
|
|
ENERXÍA
LIBRE DE REACCIÓN A
PARTIR DE ENERXÍAS LIBRES DE
FORMACIÓN
|
|
Tendo en cuenta que ΔG é unha función de estado, que só
depende dos estados inicial e final, e que temos tabulados os valores da
enerxía libre normal de formación, ΔGºf ,para diferentes
compostos, podemos calcular a súa variación nunha reacción sen máis que
sumar as enerxías libres dos reactivos e produtos que interveñen na mesma
cando ambos se atopan no estado normal, é dicir, á presión de 1atm se se
trata de gases ou á concentración de 1mol/l para substancias en disolución
líquida.
A variación de enerxía libre dunha reacción química defínese
como:
Podemos concluír que xa estamos en condicións de
determinar se unha reacción é espontánea ou non, pero é conveniente
distinguir entre espontaneidade e rapidez dunha reacción química. Que
unha reacción sexa espontánea significa que existe unha tendencia
natural para que a reacción se realice, pero poida que sexa tan lenta,
que na práctica non se aprecie ningún cambio. Así, a combustión da
gasolina é espontánea, con todo, a menos que se aplique unha faísca, a
gasolina pode manterse en contacto co aire durante longos períodos de
tempo sen que reaccione. Se unha reacción é espontánea pero lenta,
sempre é posible atopar medios para acelerar o proceso, tales como
elevar a temperatura ou empregar un catalizador. Unha reacción
espontánea é unha reacción termodinámicamente posible. Se unha reacción
non é espontánea, como a inversa da anterior (conversión de CO2
e H2O en gasolina) xamais existirá un catalizador que a faga
posible. Por tanto, o concepto de reacción espontánea ou non espontánea
permite ao químico ver os límites do posible.
EXEMPLO 6: Calcula a variación de enerxía libre na reacción seguinte en condicións estándar a partir de datos tabulados de enerxías libres de formación:
CaCO3(s) → CaO(s) + CO2(g)
Utilizando a
táboa
de datos químicos.
ΔGºR = Σ np·ΔGºfp - Σ nr·ΔGºfr =
= 1mol·ΔGºf [CaO(s)]+1mol·ΔGºf
[CO2(g)] – 1mol·ΔGºf [CaCO3(s)]=
= 1mol·(–604,2kJ/mol) +1mol·(–394,6kJ/mol) –1mol·(–1128,8kJ/mol) = 130kJ
EXERCICIOS
PARA PRACTICAR
|
|
|
|